THE TRANSCRIPTOME AND METHYLOME OF THE DEVELOPING AND AGING BRAIN AND THEIR RELATIONS TO GLIOMAS AND PSYCHOLOGICAL DISORDERS
by Henry Loeffler-Wirth, Lydia Hopp, Maria Schmidt, Roksana Zakharyan, Arsen Arakelyan and Hans Binder
ABSTRACT
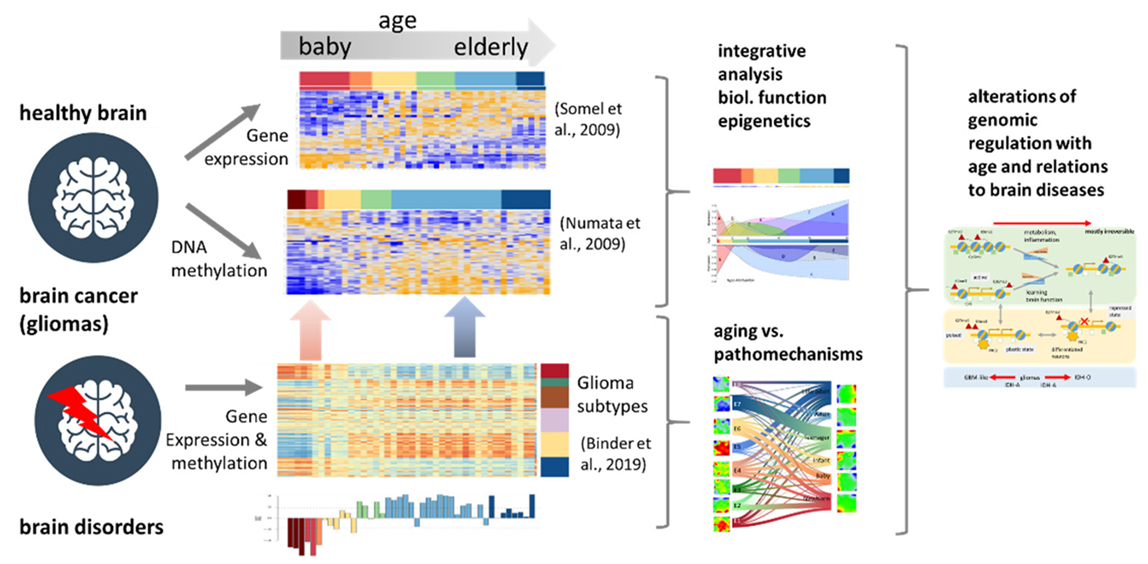
Mutually linked expression and methylation dynamics in the brain govern genome regulation over the whole lifetime with an impact on cognition, psychological disorders, and cancer. We performed a joint study of gene expression and DNA methylation of brain tissue originating from the human prefrontal cortex of individuals across the lifespan to describe changes in cellular programs and their regulation by epigenetic mechanisms. The analysis considers previous knowledge in terms of functional gene signatures and chromatin states derived from independent studies, aging profiles of a battery of chromatin modifying enzymes, and data of gliomas and neuropsychological disorders for a holistic view on the development and aging of the brain. Expression and methylation changes from babies to elderly adults decompose into different modes associated with the serial activation of (brain) developmental, learning, metabolic and inflammatory functions, where methylation in gene promoters mostly represses transcription. Expression of genes encoding methylome modifying enzymes is very diverse reflecting complex regulations during lifetime which also associates with the marked remodeling of chromatin between permissive and restrictive states. Data of brain cancer and psychotic disorders reveal footprints of pathophysiologies related to brain development and aging. Comparison of aging brains with gliomas supports the view that glioblastoma-like and astrocytoma-like tumors exhibit higher cellular plasticity activated in the developing healthy brain while oligodendrogliomas have a more stable differentiation hierarchy more resembling the aged brain. The balance and specific shifts between volatile and stable and between more irreversible and more plastic epigenomic networks govern the development and aging of healthy and diseased brain.
INTRODUCTION
The human brain relies on the lifelong function of its diverse neuronal cell types. Most neurons emerge during development in the fetal and baby’s brain and need to be maintained throughout adulthood. Neuronal health is governed by cellular programs, which need to be flexible to mediate neuronal plasticity, and yet stable to express a constant set of cell-type-specific genes throughout the life of the neurons [1]. During aging, the mechanisms that normally maintain health, stress resistance, and brain function decline, resulting in decrepitude, frailty, and the increased risk of aging-associated psychotic and neurodegenerative disorders and also of cancer [2]. The dysregulation of transcriptional and chromatin networks is a crucial component of aging [3]. Hereby, epigenetic, changes including DNA methylation, histone modifications, and chromatin remodeling, profoundly affect cellular function, thereby contributing to the progression of aging and to age-related declines in cognition. Additionally, neuronal differentiation in early life is tightly regulated by transcriptional and epigenetic mechanisms [4] with impact also for high-order cognitive functions such as learning and memory [5]. Molecular shaping during prenatal development via DNA methylation, histone modifications, and other molecular constituents of the epigenome are likely to play a critical role in the maintenance of neuronal health and function throughout the entire lifespan [6]. The dysregulation of any of these mechanisms can result in neuropsychiatric disorders, such as depression, schizophrenia, and autism spectrum disorders [2,7]. Previous studies show that gene expression and DNA-methylation dynamics are largest from birth through infancy, after which these molecular profiles transition to a relatively stable state by young adulthood [8,9]. Later in life, DNA methylation drifts with age [10] with impact on other epigenetic marks, such as histone modification, in turn affecting chromatin states [11]. Overall, altered DNA methylation is among the central mechanisms in development, aging, and cellular senescence [12].The aging and development of brain cancer are two interrelated processes, with aging being a major risk factor for the initiation of cancer. Analysis of epigenetic aging signatures of 25 cancer types revealed that DNA methylation patterns hardly reflect chronological age of cancer patients, but they are coherently modified in a non-stochastic manner, particularly at CpG (CG rich; see list of abbreviations below) islands that become hypermethylated upon aging in non-malignant tissues [13]. Brain cancer cells are diverse in their genetic, metabolic. and micro-environmental compositions, accounting for their phenotypic heterogeneity and disparate responses to therapy. These factors converge at the level of the epigenome, where aberrant epigenomes define many childhood and adult brain cancers, as demonstrated by widespread changes of DNA methylation patterns, redistribution of histone marks. and disruption of chromatin structure [14]. This coordinated regulation of cancer signatures seems to resemble aspects of aging and development.In a wider context, mutually linked expression and methylation dynamics in the brain govern regulatory mechanisms in the genome during development and aging occurring over the whole lifetime and determining the developmental stage and later the onset and the progression of aging [15]. Quantifying the extent and identity of transcriptional and epigenetic changes is therefore important for understanding development, aging. and related disorders and cancer [16]. In this publication, we have analyzed transcriptome and DNA-methylation data collected separately from human brain samples over the whole lifespan [9,17] (Figure 1). Our integrated analysis aims at systematically linking transcriptional programs and DNA-methylation changes as a function of age between early childhood and late adulthood. We decomposed molecular aging profiles into characteristic modes using self-organizing maps data portrayal, a machine learning approach proven in previous large-scale studies on transcriptome [18,19] and joint transcriptome/methylome data [20,21,22]. Hereby, we aimed to identify the functional context of the modes and their relation to epigenetic mechanisms such as the expression of genes encoding chromatin-modifying enzymes and of genes located in different chromatin states [23,24]. Finally, we compare the aging profiles of the healthy brain with expression and methylation data of brain cancer (lower-grade gliomas) and with gene signatures derived from psychotic disorders [16] in order to identify footprints of the respective pathophysiologies related to brain development and aging.
…
CONCLUSIONS
Our analysis highlighted aspects of the dynamic nature of the developing and aging epigenome and of associated cellular functions. A more systematic understanding of the role and precise timing of age-associated changes in molecular players is required for a deeper insight into these processes. One important point in the role of epigenomic changes with age is the balance between volatile and stable and between more irreversible and more plastic networks. Understanding how genetic aberrations and environmental stimuli modulate these networks is required not only to increase our understanding of aging, but also of the initiation and courses of neuropsychological disorders and of cancer. Larger-scale analyses and increased cellular resolution are needed to link molecular players such as gene expression and DNA methylation with neuronal nets and cognitive functions and their development and aging.
click here to read the full article: https://www.mdpi.com/2073-4409/11/3/362